
Speaking With Your Mind: Restoring Speech in ALS
Post by Anastasia Sares
The takeaway
In this case study, scientists demonstrate a system that can take signals from electrodes implanted in the brain and turn them into speech that can be played through a speaker. In this way, they were able to restore speech capacity to a man who had lost the ability to speak due to amyotrophic lateral sclerosis (ALS).
What's the science?
Amyotrophic Lateral Sclerosis (ALS) is a debilitating disease where motor neurons gradually atrophy and die, leaving the sufferer unable to move their bodies, though their brain continues to function normally. You may remember the “ice bucket challenge,” an ALS fundraiser that went viral on social media in 2014. Ten years later, the money raised from that challenge has done an enormous amount of good, advancing research and care, and new treatments have come to market that can slow the progression of the disease. However, ALS is still without a cure. In late-stage ALS, motor function deteriorates enough that people’s speech becomes extremely slow and distorted, which dramatically affects their quality of life.
This week in the New England Journal of Medicine, Card and colleagues published a case study of a man with advanced ALS who received brain implants that allow him to speak with the aid of a brain-computer interface.
How did they do it?
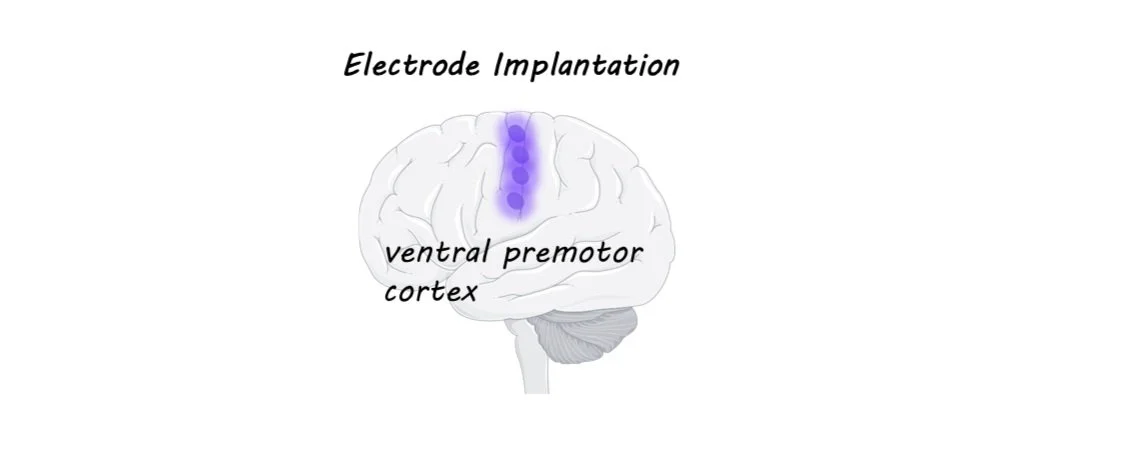
Electrode arrays have been implanted in brains before, often in patients with severe epilepsy who have to undergo brain surgery anyway in order to monitor and treat their condition. In these experiments, electrode arrays (chips with a bunch of tiny electrodes in a grid-like pattern) have been placed in various spots in the brain and scientists have been able to figure out which regions have activity that can be “decoded” to correctly predict speech. The best areas are around the ventral premotor cortex (see image).
In this study, the authors used what had been learned from previous research and chose four spots along this premotor strip to implant the electrodes in this patient. The signals from the electrodes were sent via a cable to a computer, where a neural network was used to match the brain activity with the most likely phoneme (a phoneme is a speech sound like “sh” or “a” or “ee”) that the man was trying to say. The string of phonemes was then sent to two separate language models: the first predicted possible words from the phonemes, and the second predicted possible phrases from the individual words. These models function in a similar way to the predictive text on your phone or in speech-to-text software. Finally, the predicted word sequence was turned into speech at the end of each sentence, using a synthesized voice created from the man’s own pre-ALS speech samples.
What did they find?
The authors evaluated the accuracy of the system in two ways. First, they prompted the man to think of certain words and phrases to see if the system could reliably reproduce the prompt. Second, they allowed the man to “speak” freely and then had him evaluate whether the system had faithfully produced what he wanted to say. Since the patient could not move, they had him do the evaluation using a rating screen with different bubbles (“100% correct,” “mostly correct,” and “incorrect”) and an eye-tracking system that could track which of the rating bubbles he looked at. The system started with about a 10% error rate, which gradually reduced over time as the system was trained to only 2.5% errors, with a vocabulary size of 125,000 words—a substantial increase in performance compared to the few other studies of this kind. The patient’s speaking rate also increased from the 6 words per minute he could produce naturally to around 30 words per minute (the normal English speaking rate is close to 160 words per minute).
What's the impact?
This study demonstrates how brain-computer interfaces are not only possible but can dramatically improve the quality of life for those who have lost normal functioning due to disease. As stated in the article, the first block of trials was excluded from the experiment because “the experience of using the system elicited tears of joy from the participant and his family as the words he was trying to say appeared correctly on screen.” Videos of the system can be accessed on the page of the original publication.



