
The Effects of Traumatic Brain Injury in the Presence of Aß Pathology
Post by Negar Mazloum-Farzaghi
The takeaway
There is a link between traumatic brain injury and the development of dementia. The double burden of traumatic brain injury and Aß deposits in the aging brain can cause alterations in cellular processes that lead to neuron death.
What's the science?
Previous research has established an association between traumatic brain injury (TBI) sustained in early adulthood and the risk of developing dementia later in life. It is less clear whether TBI sustained as an elderly individual (geriatric TBI) increases the risk of developing dementia, and what the cellular mechanisms would be involved. The aging brain’s response to TBI may be influenced by several factors, such as increased inflammation, increased amyloid beta load (Aß; a hallmark of dementia), and decreased autophagic activity (important process for degrading unnecessary or dysfunctional intracellular macromolecules). This week in Scientific reports, Streubel-Gallasch and colleagues used an in vitro model, designed to simulate the conditions of the aging brain, to investigate cellular responses to TBI in the presence of Aß pathology.
How did they do it?
In order to investigate the effects of physical injury caused by TBI in the presence of Aß deposits, the authors exposed co-cultures of astrocytes (specialized glial cells fundamental to maintaining homeostasis and cellular function) and neurons to Aß protofibrils. After the exposure, they subjected the cultures to a “scratch injury” using a scalpel in order to mimic TBI. There were four experimental groups: untreated (control), scratch injury (TBI), Aß protofibril exposure, and the combination of both TBI and Aß.
The authors performed immunocytochemistry to examine the effect of TBI and Aß protofibril exposure on neuronal survival. They employed the same method to examine how TBI and Aß protofibril exposure affects astrocytes. To do this, they used two well characterized astrocytic markers, Glial fibrillary acidic protein (GFAP) and calcium-binding protein (S100ß). They used transmission electron microscopy to further assess changes to cellular homeostasis caused by TBI and Aß protofibril exposure by examining the function of the mitochondrial network of astrocytes. Finally, using both immunocytochemistry and immunoblotting, they also examined whether autophagic activity was impaired in response to TBI and Aß protofibril exposure.
What did they find?
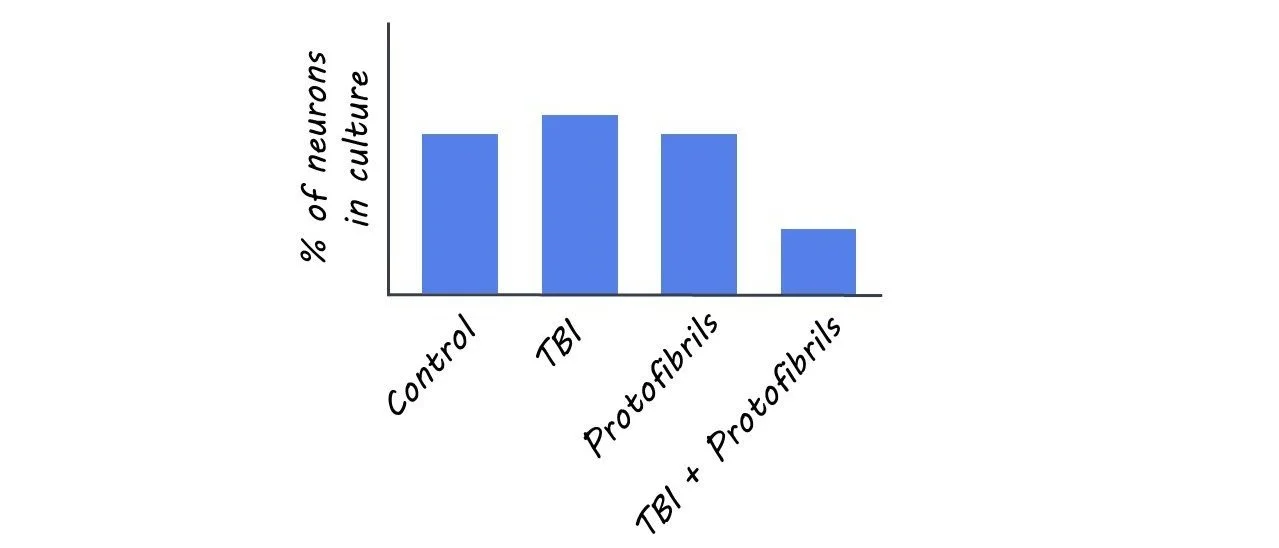
The number of neurons in the experimental TBI group, Aß protofibril-exposure group, and control group remained stable. However, in the experimental group with the combination of Aß and TBI, the percentage of neurons decreased significantly. One explanation for this finding may be that the double burden of Aß deposits and TBI hindered astrocytic functions, which threw their ability to maintain homeostasis off balance, making it more difficult to clear debris and protect neurons.
Aß protofibril-exposed astrocytes displayed higher GFAP levels. In contrast, astrocytes subjected to both Aß and TBI displayed lower GFAP levels. This was a surprising finding as GFAP usually upregulates in response to TBI. The other astrocytic protein, S100ß, showed increased levels in astrocytes in all three experimental groups compared to the control group. This is in line with previous work that has shown that S100ß upregulates in response to trauma and has increased levels in the tissue cells and astrocytes of dementia patients. However, it remains to be determined whether these increased levels have positive or negative effects in pathological conditions.
Aß pathology and TBI caused mitochondrial damage in astrocytes (but no clear additive effect of the two), which may have negative consequences for vital astrocytic functions thereby disturbing homeostasis. There was an increase in autophagic activity in the Aß protofibrils exposure group and TBI group. Interestingly, when cells were exposed to a combination of Aß protofibrils and TBI, autophagy failed to upregulate, which may be indicative of an inefficient compensatory mechanism when faced with this double burden.
What's the impact?
This is the first study to show that the double burden of Aß deposits and TBI result in neuronal loss, altered astrocytic responses as indicated by changes in the expression of two key astrocytic proteins, mitochondrial disturbances, and abnormal autophagic activity. The findings of this study demonstrate the underlying cellular mechanisms involved in the relationship between TBI and the development of dementia. Overall, this study advances our understanding of geriatric TBI on the cellular level.
