
Early Intervention in Huntington’s Disease Mutation Carriers Delays Symptom Onset
Post by Andrew Vo
The takeaway
Genetic mutations that cause Huntington’s disease later in life also affect neurodevelopment in early life. Medications that enhance the glutamate neurotransmitter system in mutation carriers during infancy have the potential to prevent the later onset of disease.
What's the science?
Huntington’s disease (HD) is a neurodegenerative disease that is largely inherited through mutations in the huntingtin gene (HTT). HD symptoms do not become apparent until middle or late adulthood, but the underlying mutation has been shown to affect brain development much earlier in life. Although the brain can compensate for this early dysfunction, it is unknown whether intervening during this period can delay or prevent the later development of disease. This week in Science, Braz et al. use a mouse model of HD to demonstrate how treating early brain abnormalities in mutation carriers can alter the disease course.
How did they do it?
First, the authors recorded activity from neurons of newly born mice expressing the HTT mutation (i.e., HD mice). Similarly, they conducted brain recordings in postnatal mice depleted of HTT (i.e., HTT-depleted mice). Together, these experiments investigated the HTT gene's critical role in HD development. Next, the authors investigated whether enhancing excitatory neurotransmission affected both early brain physiology and the later onset of HD symptoms. Postnatal HD mice and non-mutant control mice were treated with CX516, which increases glutamate neurotransmitter signalling, and the effects on behavioural symptoms were observed during adolescence and adulthood. The effects of HTT mutation and CX516 treatment on brain structure were also measured using magnetic resonance imaging.
What did they find?
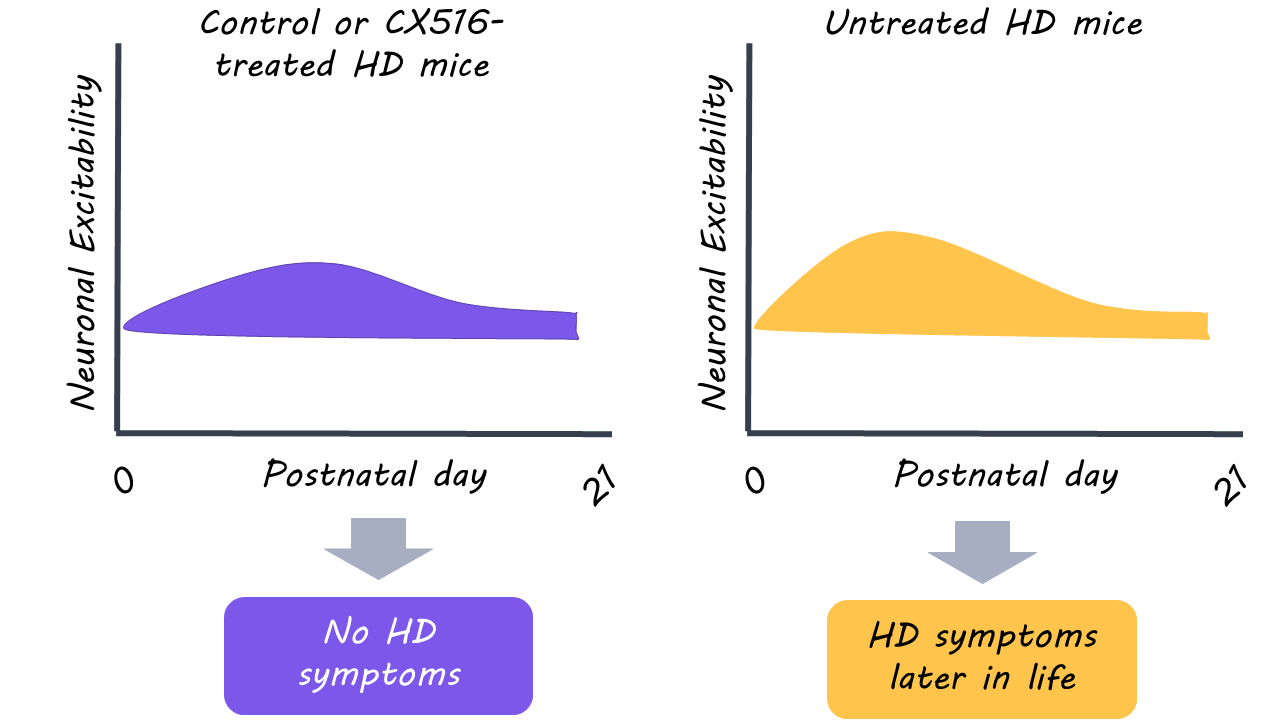
During their first week, postnatal HD mice showed evidence of reduced excitatory activity and less complex neurons compared to non-mutant mice. By the second week, however, the brains of the HD mice were able to normalize. Postnatal HTT-depleted mice displayed similar defects in neuronal activity as HD mice, however, these abnormalities failed to normalize. The results suggest that at least partial HTT gene function is critical for the brain to compensate against early HD dysfunction.
Administration of CX516 in postnatal HD mice restored neuron shape and motor function in comparison to untreated HD mice. Astonishingly, this postnatal treatment of excitatory glutamate signalling prevented the later development of HD symptoms in adulthood. Early intervention also appeared to normalize brain structure (as measured with magnetic resonance imaging) in adult HD mice; brain volume was reduced in untreated HD mice but not in those with early intervention. It should be noted, however, that CX516 had negative effects in non-mutant mice. This indicates that there is an optimal range of excitatory neurotransmission for normal brain function.
What's the impact?
This study demonstrated that treatment of early brain dysfunction in HD mutation carriers might delay or prevent the onset of disease. Although this was achieved in a mouse model of HD, it nonetheless represents an important step toward a cure for human patients.
